Quantitative Structure-Activity Relation Study of Quaternary Ammonium Compounds in Pathogen Control: Computational Methods for the Discovery of Food Antimicrobials
Ethan C Rath and Yongsheng Bai
DOI10.21767/2470-6973.100017
Ethan C Rath1 and Yongsheng Bai1,2*
1Department of Biology, Indiana State University, Terre Haute, Indiana, USA
2The Center for Genomic Advocacy, Indiana State University, Terre Haute, Indiana, USA
- *Corresponding Author:
- Yongsheng Bai
Department of Biology and The Center for Genomic Advocacy
Indiana State University Terre Haute
Indiana, USA
E-mail: Yongsheng.Bai@indstate.edu
Received date: May 27, 2016; Accepted date: June 20, 2016; Published date: June 25, 2016
Citation: Rath EC, Bai Y. Quantitative Structure-Activity Relation Study of Quaternary Ammonium Compounds in Pathogen Control: Computational Methods for the Discovery of Food Antimicrobials. Chem Inform. 2016, 2:1.doi: 10.21767/2470-6973.100017
Abstract
Objective: Quaternary ammonium compounds (QACs) are surfactants that are made of at least one cationic nitrogen attached to a variety of different side groups, usually consisting of one or more hydrophobic chains. These compounds are generally used for surface decontamination, oral hygiene, and recently in carcass preservation. Recently there have been many studies that have implicated QACs in the development of resistance in bacteria as well as harmful environmental effects. One compound in particular, cetylpyridinium chloride (CPC), has recently gained acceptance as a safe and practical method for use in consumable raw poultry product decontamination. This compound is highly lipophilic and leaves a residue that is potentially toxic to consumers and the environment if not properly removed.
Methods: Using computational methods, we propose the use of quantitative structure-activity relation (QSAR) analysis to determine the antimicrobial effects of novel and untested QACs and QAC-like, structures for further testing.
Results: We developed a consensus model with an R2 and a slope of 0.98, which shows good linear structure of its predictions of minimum inhibitory concentration (MIC). This model was validated by prediction of known antimicrobial data of QACs. Similar compounds to CPC were collected and their antimicrobial effects were predicted by this model. Many of these compounds were detected as possible antimicrobials.
Conclusion: This study has identified several promising antimicrobial compounds worth of further study. By diversifying the available QACs we hope to develop better disinfectants, create more environmentally friendly compounds, and help to stall, or even halt, the development of antimicrobial resistance.
https://transplanthair.istanbul
https://hairclinicturkey.co
https://hairclinicistanbul.co
https://besthairtransplant.co
https://hairtransplantistanbul.co
Keywords
Quantitative structure activity relations; Chemoinformatics; Disinfectant; Cetylpyridinium chloride; Quaternary ammonium compounds; Antimicrobial; Minimum inhibitory concentration; Computational chemistry; Surfactant
Background
Bacterial infection on the surface of fresh meats and produce after processing is currently one of the largest problems within this industry. Bacteria that cause most foodborne illness and include, but are not limited to, shiga toxin producing Escherichia coli (E. coli) and Salmonella typhimurium (S. typhimurium) [1,2]. Not only do these bacteria cause disease, but also spoilage. It is estimated that in 2010 the United States of America threw out 133 billion pounds of food, mostly due to spoilage [3]. These bacteria cannot be removed by simple water spraying implemented by most processing facilities [4]. As such, many technologies have been developed to combat bacteria on the surface of food products. These technologies involve the use of chlorine, chlorine dioxide, Salimide, ozone, and cetylpyridinium chloride [5-9].
Unfortunately, the current technologies being used to remove bacteria from these surfaces suffer from a variety of issues: high cost, hazardous byproducts, environmental hazards, and the discoloration of products [9,10]. This study focuses on the use of cetylpyridinium chloride (CPC) for decontamination. CPC is an effective antimicrobial, it has been approved only for use on raw chicken, although it has also shown effectiveness for use on beef and produce for both disinfection and the extension of shelf life [11-13]. CPC is classified as a quaternary ammonium compound (QAC), which is defined by its cationic nitrogen head. Generally QACs work as antimicrobials by disrupting cell walls and membranes with hydrophobic tails. These tails pinch off sections in small vesicle-like structures and cause cell leakage that eventually leads to cell death [14-16]. CPC follows this same mechanism along with evidence of other more specific targets including transferrin denaturation, ionic channel blocking, and knock-down of halitosis specific transcription factors [14,17,18].
It does, however, have its own flaws. CPC is an environmental hazard and leaves a toxic residue on surfaces [13,19]. This residue is dissolved and subsequently removed using propylene glycol (PEG) as a cosolvent with water. Unfortunately, this adds to the cost and complicates the safe disposal of CPC [20]. Environmentally, the disposal of CPC is a major concerning factor. CPC is naturally broken down by bacteria, but in higher concentrations it kills the bacteria before it can be processed. In aquatic environments residual CPC causes a decrease in microflora and in algae blooms. This decrease causes a trophic cascade, negatively impacting all organisms in the local community [21]. The remnants of CPC in the environment can also propagate antimicrobial resistance in the local microbial communities, which can also have a lasting impact [22]. In humans, QACs taken orally in high doses (100- 400 mg/kg) have shown detrimental effects including mucosal necrosis, hemorrhaging, formation of ulcers, and severe liver, kidney, and heart changes [23,24]. CPC in particular has been shown to cause liver and kidney vacuolization as well as paralysis when given orally to rats and rabbits [25].
Discovery of novel drugs is typically limited by the funds available and the precise knowledge of drug targets. Due to the nonspecific nature of CPC and imprecise library screening methods, our lab turned to qualitative structure activity relationships (QSAR). QSAR allows for the recoding of molecular structures to quantifiable forms which are then correlated to a specific biological activity. This model can then be used to predict the biological activity of untested structures [26]. The bioactivity that we wish to study is the minimum inhibitory concentration (MIC), which is a measure of the effectiveness of an antimicrobial. A lower MIC denotes a more effective compound. Using this method we hope to discover potential structures that could function as well as CPC, with reduced or nonexistent negative effects on the human body and the environment.
Methods
Data collection
Three sections of data were collected via literature searches (1) a model building set, (2) a validation set, and (3) a prediction set of compounds [27-29]. The model building set was based on known QACs with data on the MIC of these compounds against E. coli. Contained within the validation set were known QACs that were not used for the model building set. Compounds for the prediction set were collected from a substructure search on Pubchem using CPC as a reference. The top 1000 compounds sorted by relevance were selected for further testing.
Descriptor calculation
All descriptors for the model building set (Supplemental Data 1), the validation set (Supplemental Data 2), and the prediction set (Supplemental Data 3) were calculated simultaneously using the ochem.eu chemical database [30]. Using the tools on this site, the structures were cleaned by removing the salts associated with each compound. Under the models tab, calculate descriptors program was selected and the SMILES string for each compound was uploaded in an Excel file (.xls). These SMILES were used to calculate descriptors through this database. The descriptors that were selected are the following: E-state (all but extended indices), ALogPS, GSFragments, ISIDA fragments (from 2-15 in order to cover long carbon chains), and QNPR. These were selected due to a large number of compounds encountering errors during 3D structure calculations. Unless noted, all descriptors were left at the default settings. This totaled 1356 descriptors for each compound. The descriptors and the chemID’s were then downloaded as a .cvs file ignoring any compounds that encountered an error. The model building set and the validation set had no errors and 163 compounds were removed from the prediction set due to errors in calculation.
Data preprocessing
After the descriptors were calculated, all data were normalized through the Normalize Data (v.1.0) tool developed by the Roy lab [31]. This is a Java program that requires a .csv file of the descriptors. The model building set data was then split into a test (15%) and training set (85%) via the Data set Division GUI (v.1.2) also developed by the Roy labs [32,33] (https://teqip.jdvu.ac.in/ QSAR_Tools/#ADInHouse).
QSARINS model calculation
Using QSARINS, an open source QSAR modeling software utilizing multiple linear regression (MLR), was used to create the QSAR model and to generate each prediction [34,35]. First, the model building set was altered to fit the QSARINS format. The MIC was then added to the descriptors column and the test and training sets were combined into a single file where each was given a numerical identifier (1 for training set, 2 for test set) in the last column of the file. This was saved as a .txt file. The software was run according to the protocol listed in the manual. We used their internal filters to remove all descriptors that had <80% consistency throughout the data set, or that were <95% correlated. The genetic algorithm was run for combinations of up to 130 descriptors based on the Q2 loo. 840 models were created, using QSARINS available validation data. An arbitrary cutoff of R2>0.75, R2-Q2<0.10 (both loss of one and loss of many), and |Q2 - Y-scramble| > 0.50 was used. Twelve models were left for further validation. Predictions for the prediction set and the validation set were performed using the built-in tool. (https://www.qsar.it/).
Results
Model validation
In order to find a new chemical to treat meat surfaces, we performed a literature search for current QACs and their respective MIC against E. coli [27-29]. The compounds that we found had at least one cationic nitrogen and a carbon chain. Other commonly identified structures include nitrogen, oxygen, benzene rings, and even barium in one compound. Activities of these compounds range from an MIC of 1.88 μg/ml to 12800 μg/ml. Using all available literature data on the antimicrobial activity of currently available QACs on E. coli represented by the log of the MIC, we developed 840 potential models using the QSARINS software. QSARINS systematically uses optimized descriptors to build models starting at 1 descriptor and building more complex models using a genetic algorithm (GA). The GA organizes the descriptors into genes in a chromosome and then other descriptors are substituted into this chromosome. This continues with a constant mutation rate for 500 generations. At the end of these generations each chromosome is used to create an MLR based QSAR model. The top five models (determined by the Q2loo) are kept for each iteration. The number of descriptors is increased as time progresses and more calculations are done. Due to computing limitations, this process was stopped at 130 descriptors, although most optimal models had fewer than eight descriptors. The top models had some descriptors in common, or at least very similar fragments. The H-C-O structure fragment was seen in 10 of the top 12 models. We organized these descriptors into four categories to explain the importance of certain types of descriptors for this model calculation: (1) short fragments (specific fragments of five atoms or less), (2) long fragments (specific fragments of more than five atoms), (3) non-specific fragments (fragments with general patterns and not specific structural identities, examples include C*C*N:(Fragmentor) in which “*” could be any atom), and (4) log of the lipophilicity which was calculated by A*log(PS) (Table 1).
| Model ID | Variables |
|---|---|
| Short Fragments | 20 |
| Long Fragments | 10 |
| Non-specific Fragments | 20 |
| Log(lipophilicity) | 4 |
Table 1: Classification of Descriptors used in prediction calculation.
In order to select the best potential models from the 840 potential models, a general filter of R2>0.75, R2 - Q2<0.10, and |Q2 - Y-scramble| >0.50 was used to reduce the list to 12 potential models based on internal validation calculations done with the QSARINS software (Table 2). The majority of compounds were removed due to the R2 - Q2 filter. An external testing dataset was then predicted by the model in order to perform an external validation. For this study we focused on the general prediction ranking (R2) and the specific accuracy of our prediction (percent error). These were calculated and are displayed in Table 3. It is typical in the QSAR community to rely more on the general predictive ranking than to rely on accuracy alone, as these predictions will be used for filtering a larger list for experimental validation rather than for direct prediction [36]. Many of the models were very similar in their validations, therefore the most optimal model, 81, was selected to provide an example of the internal and external regressions (Figures 1 and 2).
| Model ID | Variables | R2 | R2-Q2loo |
|---|---|---|---|
| 94 | 7 | 0.838 | 0.0802 |
| 82 | 5 | 0.8116 | 0.0659 |
| 81 | 5 | 0.8024 | 0.0713 |
| 72 | 4 | 0.7933 | 0.0741 |
| 70 | 4 | 0.7929 | 0.0813 |
| 75 | 5 | 0.789 | 0.079 |
| 69 | 4 | 0.7877 | 0.0816 |
| 67 | 4 | 0.7656 | 0.0713 |
| 63 | 4 | 0.7651 | 0.0765 |
| 66 | 4 | 0.76 | 0.0681 |
| 64 | 4 | 0.7585 | 0.0685 |
| 65 | 4 | 0.7576 | 0.0675 |
Table 2: Internal Validation of Select 12 QSAR models.
| Model ID | Average % Error | R2 |
|---|---|---|
| 94 | 14% | 0.8917 |
| 82 | 14% | 0.9274 |
| 81 | 11% | 0.9578 |
| 72 | 24% | 0.8804 |
| 70 | 22% | 0.8828 |
| 75 | 19% | 0.8566 |
| 69 | 25% | 0.8081 |
| 67 | 18% | 0.9101 |
| 63 | 16% | 0.9242 |
| 66 | 20% | 0.8772 |
| 64 | 14% | 0.9254 |
| 65 | 72% | 0.1967 |
| Consensus (all) | 9% | 0.971 |
| Consensus (selected) | 10% | 0.9439 |
| Consensus (worst removed) | 13% | 0.9315 |
Table 3: External Validation of Select 13 QSAR models.

Figure 1: Flow chart of the QSAR building process, with software used at each step.

Figure 2: The top QSAR model based on the R2 values for the predictions of the external validation set. A) Model 81 training and prediction set regression, the training set is in yellow and the prediction is in blue. B) Model 81 external validation regression, the R2 is displayed on the graph.
Many studies have pointed to the effectiveness of using a consensus model for increasing the accuracy of the prediction of unknown models, rather than using a single model [36,37]. Using the twelve previously identified models, we averaged the predictions on the validation set to develop three different consensus models (Table 2 and Figure 3). One model was created from all available models. The second was made by selected models that had a R2>0.9 and an average error <20%. The third consensus was formed by removing model 65. This model had the worst external validations with an R2 of 0.19 and an average error of 72%. These consensus models generally had lower error and higher R2 than the single models. The removal of lesser models or the single worst model did not improve the accuracy of the consensus. From the validation data we determined that the consensus model made from all the available models, as previously described, would be the preliminary optimized model to use for predictions of unknown compounds.
Figure 3: Predictions on an external validation, regressions of three different consensus models. In green, all available models that met our cutoffs, the blue is all models that had an external validation R2≥ 0.90 and an average standard error ≤ 0.20, the yellow is a regression with the single worst model (having an R2 of 0.19). Each R2 is displayed under the legend heading for each data set.
Predictions for unknown compounds
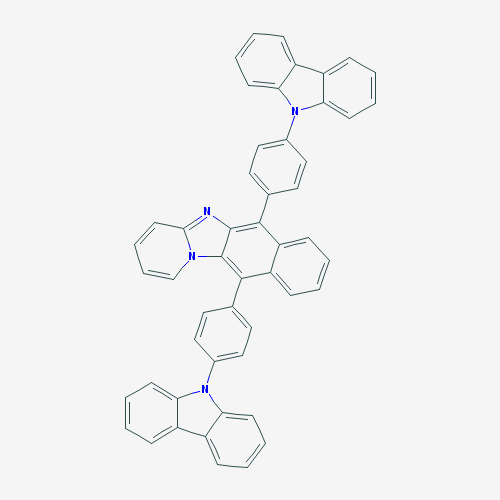
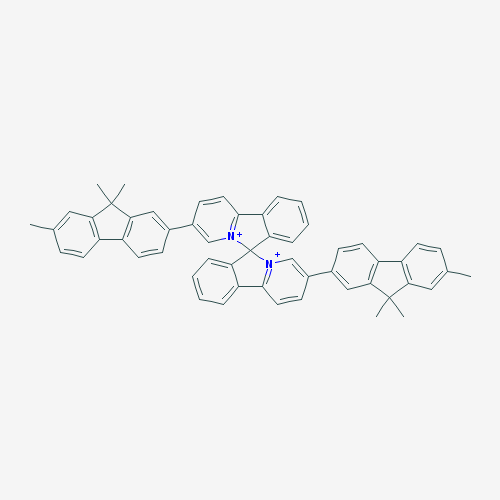
The purpose of creating a QSAR model is to apply it to previously unstudied compounds with unknown biological activities. We collected a list of 1000 compounds from PubChem that had substructure similar to CPC [38]. By using the consensus model, the top 10 compounds in terms of MIC against E. coli were identified. Compounds that were in the applicability domain for at least 75% of the models within the consensus were included in the final list. This left us with 39 compounds. These compounds, their structures, and their predicted activities are shown in Table 4.
| Compound | Predicted MIC (μg/ml) | Compound | Predicted MIC (μg/ml) |
|---|---|---|---|
 |
0.969267 | 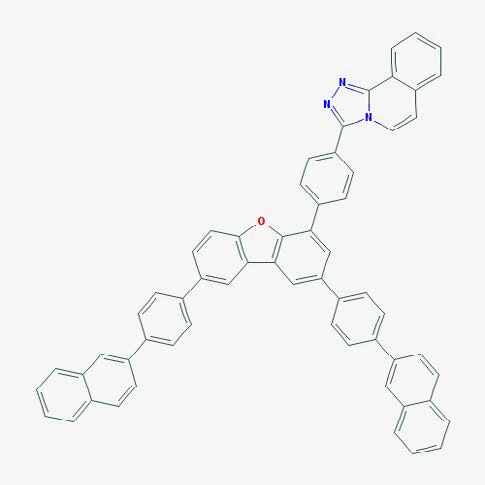 |
1.077558 |
 |
1.016167 | 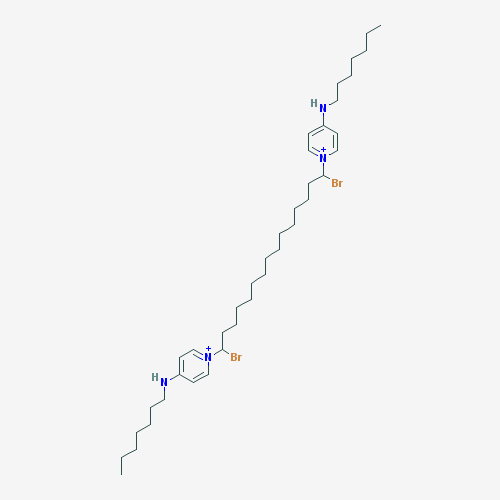 |
1.083292 |
 |
1.016167 | 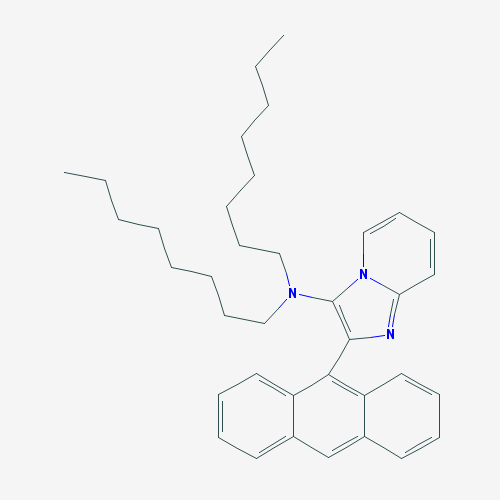 |
1.097142 |
 |
1.043917 | 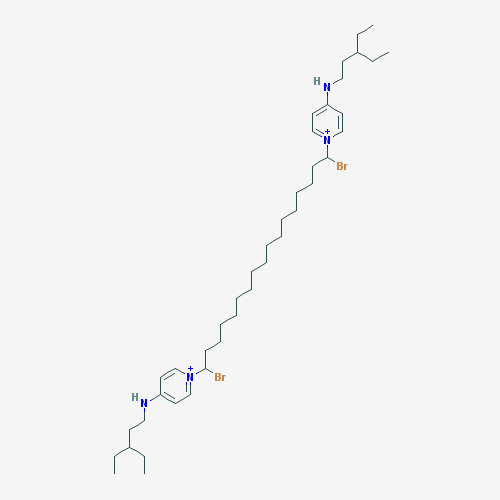 |
1.101175 |
 |
1.071908 | 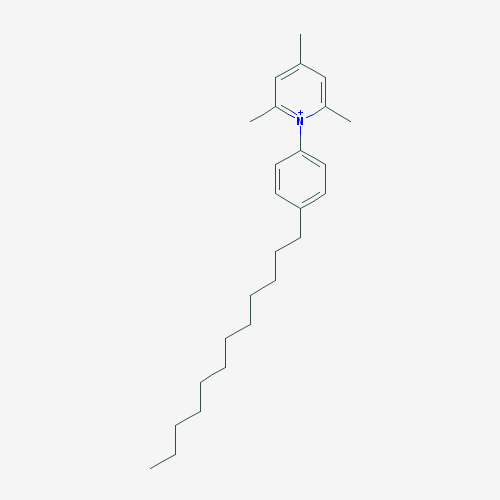 |
1.118575 |
Table 4: Top 10 potential compounds, determined by predicted MIC.
Discussion
Using literature values, a QSAR model was developed in order to predict the MIC of potential compounds that could be used to combat bacteria on the surface of food during processing. Our model was based on 47 compounds with available literature values with recorded MIC values against E. coli, collected across three different studies to increase the variation of structures and MIC values. Using the built in GA the best descriptors and the optimal number of descriptors were selected to avoid overtraining of the model. Some may argue that only using up to 130 descriptors could be a detriment to our study but, any calculations done with more than 15 variables there was a significant decrease in Q2 leading us to believe that overtraining had occurred beyond that point.
Now that we have a viable QSAR model of MIC and preliminary predictions for almost 900 structures, we plan to experimentally validate the predicted MIC. After this validation our lab will focus on creating two more models 1) one to predict the environmental degradation of these compounds and 2) one that would predict the amount of residue that would be left on different food products when the compounds are used for sterilization. These steps will help us to discover a safer compound from the list of potential compounds.
Disinfectants in the food industry are incredibly important for the reduction of spoilage causing bacteria as well as those that can cause disease. Unfortunately, current techniques have many issues. One compound that is efficient in both cost and in antibacterial action is CPC, but the remaining residue must be removed or the products could become toxic. In order to find a comparable compound without the toxic residue, our lab developed a QSAR model that could predict the antimicrobial activity of potential compounds before experimental testing. This model will allow us and other labs to save money and time by specifically testing compounds that have predicted efficacy for antimicrobial behavior. By developing and testing new antimicrobial QACs we hope to not only reduce the bacteria on the surface of food in a safe manner, but also reduce the amount of antimicrobial damage to the local environment. With the addition of new QACs we also expect to help combat the rise in antibiotic/antimicrobial resistant bacteria.
Acknowledgements
Our lab would like to thank Hunter Gill for his contributions to this project and Dr. Gary Stuart and Stephanie Pitman for their comments and edits.
References
- Berends BR, Van KF, Mossel DA, Burt SA, Snijders JM (1998) Impact on human health of Salmonella spp. on pork in The Netherlands and the anticipated effects of some currently proposed control strategies. Int J Food Microbiol 44: 219-229.
- Erickson MC, Doyle MP (2007) Food as a vehicle for transmission of Shiga toxin-producing Escherichia coli. J Food Prot 70: 2426-2449.
- Buzby JC, Wells HF, Hyman J (2014) The Estimated Amount, Value and Calories of Postharvest Food Losses at the Retail and Consumer Levels in the United States. Retrieved from https://www.ers.usda.gov/publications/eib-economic-information-bulletin/eib121.aspx
- Bacon RT, Belk KE, Sofos JN, Clayton RP, Reagan JO, et al. (2000) Microbial populations on animal hides and beef carcasses at different stages of slaughter in plants employing multiple-sequential interventions for decontamination. J Food Prot 63: 1080-1086.
- Cutter C, Warren D, Handie A, Rodriguez-Morales S, Zhou X, et al. (2000) Antimicrobial Activity of Cetylpuridinium Chloride Washes against Pathogenic Bacteria on Beef Surfaces. J Food Prot 63: 593-600.
- Mullerat J, Klapes NA, Sheldon BW (1994) Efficacy of Salmide(R), a Sodium Chlorite-Based Oxy-Halogen Disinfectant, to Inactivate Bacterial Pathogens and Extend Shelf-Life of Broiler Carcasses. J Food Prot 57: 596-603.
- Singh N, Singh RK, Bhunia A K, Stroshine RL (2002) Efficacy of chlorine dioxide, ozone, and thyme essential oil or a sequential washing in killing Escherichia coli O157:H7 on lettuce and baby carrots. Lebensmittel-Wissenschaft Und-Technologie-Food Science and Technology 35: 720-729.
- Tsai LS, Schade JE, Molyneux BT (1992) Chlorination of Poultry Chiller Water - Chlorine Demand and Disinfection Efficiency. Poultry Science 71: 188-196.
- Wabeck CJ (1994) Methods to reduce microorganisms on poultry. Broiler Industry 57: 34-42.
- Rule KL, Ebbett VR, Vikesland PJ (2005) Formation of chloroform and chlorinated organics by free-chlorine-mediated oxidation of triclosan. Environ SciTechnol 39: 3176-3185.
- Bai Y, Coleman K, Waldroup A (2007) Effect of Cetylpyridinium Chloride (Cecure CPC Antimicrobial) on the Refrigerated Shelf Life of Fresh Boneless, Skinless Broiler Thigh Meat. International Journal of Poultry Science 6: 91-94.
- Gilbert C, Bai Y, Jiang H (2015) Microbial Evaluation of Cecure-Treated (Post-Chill) Raw Poultry Carcasses and Cut-up Parts in Four Commercial Broiler Processing Facilities. Int J PoultSci 14: 120-126.
- Rodriguez-Morales S, Zhou X, Salari H, Castillo R, Breen PJ, et al. (2005) Liquid chromatography determination of residue levels on apples treated with cetylpyridinium chloride. J Chromatogr A 1062: 285-289.
- Gilbert P, Moore LE (2005) Cationic antiseptics: diversity of action under a common epithet. J ApplMicrobiol 99: 703-715.
- Ioannou C, Hanlon G, Deyner S (2007) Action of Disinfectant Quaternary Ammonium Compounds against Staphylococcus aureus. Antimicrob Agents Chemother 51: 296-306.
- Wessels S, Ingmer H (2013) Modes of action of three disinfectant active substances: A review. RegulToxicolPharmacol 67: 456-467.
- Liu J, Ling JQ, Wu CD (2013) Cetylpyridinium chloride suppresses gene expression associated with halitosis. Arch Oral Biol 58: 1686-1691.
- Taheri-Kafrani A, Rastegari AA, Bordbar AK (2014) The unfolding process of apo-human serum transferrin in the presence of cetylpyridinium chloride: an isothermal titration calorimetry study. ActaChimSlov 61: 645-649.
- Zhou X, Handie A, Salari H, Fifer EK, Breen PJ, et al. (1999) High-performance liquid chromatography determination of residue levels on chicken carcasses treated with cetylpyridinium chloride. J ChromatogrB Biomed SciAppl 728: 273-277.
- Kwak KY, Nakata Y (1999) Japan Patent. P. Corp.
- Tezel U, Pavlostathis SG (2015) Quaternary ammonium disinfectants: microbial adaptation, degradation and ecology. CurrOpinBiotechnol 33: 296-304.
- Buffett-Bataillon S, Tattevin O, Bonnaure-Mallet M, Jolivet-Gougeon A (2002) Emergence of resistance to antibacterial agents: the role of quaternary ammonium compounds- a critical review. Int J Antimicrob Agents 39: 381-389.
- Cutler RA, Drobeck HP (1970) Toxicology of Cationic Surfactants. Volume 4. New York: Marcel Dekker, Inc., USA.
- Gosselin RE, Smith RP, Hodge HC (1984) Clinical Toxicology of Commercial Products. 5thedn. Balitmore: Williams and Wilkins.
- Warren MR, Becker TJ, Marsh DG, Shelton RS (1942) Pharmacological and Tocicological studies on cetylpiridinium chloride, a new germicide. J Pharm ExpTher 74: 401-408.
- Silverman K (2004) The organic chemistry of drug design and drug action. 2ndedn. Elsevier.
- Cook GK, McDonald JH 3rd, Alborn W Jr, Boyd DB, Eudaly JA, et al. (1989) 3-Quaternary ammonium 1-carba-1-dethiacephems. J Med Chem 32: 2442-2450.
- Thorsteinsson T, Masson M, Kristinsson KG, Hjalmarsdottir MA, Hilmarsson H, et al. (2003) Soft antimicrobial agents: synthesis and activity of labile environmentally friendly long chain quaternary ammonium compounds. J Med Chem 46: 4173-4181.
- Zhang Y, Li G, Liu M, You X, Feng L, et al. (2011) Synthesis and in vitro antibacterial activity of 7-(3-alkoxyimino-5-amino/methylaminopiperidin-1-yl)fluoroquinolone derivatives. Bioorg Med ChemLett 21: 928-931.
- Sushko I, Novotarskyi S, Korner R, Pandey AK, Rupp M, et al. (2011) Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J Comput Aided Mol Des 25: 533-554.
- Mohamad IB, Usman D (2013) Standardization and Its Effects on K-Means Clustering Algorithm. Research Journal of Applied Sciences, Engineering and Technology 6: 3299-3303.
- Kennard RW, Stone LA (1969) Computer Aided Design of Experiments. Technometrics 11: 137-148.
- Martin TM, Harten P, Young DM, Muratov EN, Golbraikh A, et al. (2012) Does rational selection of training and test sets improve the outcome of QSAR modeling? J ChemInf Model 52: 2570-2578.
- Gramatica P, Cassani S, Chirico N (2014) QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J ComputChem 35: 1036-1044.
- Gramatica P, Chirico N, Papa E, Cassani S, Kovarich S (2013) QSARINS: A new software for the development, analysis and validation of QSAR MLR models. J ComputChem 34: 2121-2132.
- Gramatica P, Pilutti P, Papa E (2004) Validated QSAR prediction of OH tropospheric degradation of VOCs: splitting into training-test sets and consensus modeling. J ChemInfComputSci 44: 1794-1802.
- Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, et al. (2008) Combinatorial QSAR Modeling of Chemical Toxicants Tested against Tetrahymenapyriformis. J ChemInf Model 48: 766-784.
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, et al. (2016) PubChem Substance and Compound databases. Nucleic Acids Res 44: D1202-1213.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences