Screening Constitutional Isomers of C3H2O2 for Thermodynamic Properties Obtained from Systematic Electron Structure Calculations
Ramkissoon C, Szorad JJ, Ragyanszki A, Gerlei KZ, Fiser B, Szori M, Csizmadia IG, Knak Jensen SJ, Viskolcz B
Ramkissoon C1, Szórád JJ2, Rágyanszki A2, Gerlei KZ2, Fiser B2,3, Sz?ri M2, Csizmadia IG1,2, Knak Jensen SJ4*, Viskolcz B2
Department of Chemistry, University of Toronto, M5S 3H6 Toronto, Ontario, Canada
Department of Chemical Informatics, University of Szeged, Boldogasszony sgt. 6., H-6725 Szeged, Hungary
Univesidad de Pais Vasco, Av. de Tolosa, 54, E-20018 Donostia, G¡puzkoa, Guipúzcoa, Spain
Department of Chemistry, Langelandsgade 140, Aarhus University, DK-8000 Aarhus C, Denmark
- *Corresponding Author:
- Knak Jensen SJ
Department of Chemistry, Langelandsgade 140, Aarhus University
DK-8000 Aarhus C
Denmark
Tel: +(45) 87155926
Fax: +(45) 8619 6199
E-mail: kemskj@chem.au.dk
Received date: August 18, 2015; Accepted date: September 22, 2015; Published date: September 24, 2015
Citation: Ramkissoon C, Szórád JJ, Rágyanszki A, et al. Screening Constitutional Isomers of C3H2O2 for Thermodynamic Properties Obtained from Systematic Electron Structure Calculations. Chem Inform. 2015, 1:2.
Abstract
Propiolic acid is widely employed in the synthesis and preparation of many reagents, like bis-alkynes, which has revealed cytotoxicity against B16 melanoma cancer cells. In addition, propiolic acid has antifungal and antimicrobial properties. These properties have inspired us to explore the thermodynamic properties of the isomers of propiolic acid, C3H2O2, using electron structure calculations. In all 35 isomers were studied. The thermodynamic data can be useful in the pharmaceutical and medicinal industries.
Keywords
Bis-alkynes; Molecular isomers; Geometries; Transition state
Introduction
Propiolic acid is an acetylenic compound with derivatives that may be used in different organic synthesis [1]. Acetylenic compounds can be isolated from plants, fungi and insects [2]. Some have antimicrobial and antifungal properties, which are used by organisms such as the solider beetle, Chaudiognathus leconteri, as a defensive mechanism against its predators [3]. There is ongoing research that focuses on the potential of acetylenic fatty acids as the forefronts in generating new topical treatments for fungal infections [4].
Propiolic acid is also used in the preparation of bis-alkynes which has revealed noticeable cytotoxicity against certain types of cancer cells such as the B16 melanoma cancer cells [5]. Computational techniques have been used to predict molecular properties of propiolic acid [6] and in the elucidation of reaction mechanism for decarboxylation of various carboxylic acids [7]. Relevant properties of propiolic acid in industrial context are found in ref. [8,9].
Traditional research in organic chemistry started with the synthesis of a given molecule. Subsequently its various properties, like thermodynamic stability, were determined experimentally. With the arrival of high-performance computers and with the associated software development one can generate all possible isomeric structures computationally via combinatorial chemistry. This allows one to construct virtual libraries of molecular isomers. Quantum chemical molecular computations make it possible to determine the thermodynamic stability of each of these isomers even before one would attempt their synthesis in the laboratory. Various areas of non-accessible regions of chemical research would benefit from this new approach, such as drug-design and the study of molecular evolution to mention the two most obvious aspects of "molecular design". In other words this new approach can help us to study large number of molecular isomers in a variety of areas of organic chemistry.
The present study starts with a very small molecular composition of C3H2O2 that has only a relatively few isomers. In this case it is easier to demonstrate the various principles of this method of research.
Methods
All of the isomers for C3H2O2 with a singlet electronic state were generated using valence restrictions and geometry parameters from the MM2 molecular mechanics program. The hydrogen, oxygen and carbon atoms were considered to form bond with 1, 2 and 4 valences, respectively. The C3H2O2 composition could occur either as a single molecule (29 cases) or as a molecular complex formed from two of its fragments (6 cases). Complexes with more than two fragments were not found. Based on graph theory Molgen 5.0 program [10] was employed to generate all possible singlet state initial structures using the above mentioned restrictions. These structures were used as the primary structures to determine the local minima of the potential energy surface of C3H2O2. Geometries were optimized at the B3LYP/6-31G(d) level of theory. The maximum optimization step size was restricted to 0.01 Bohr and the maximum number of the optimization steps were set to 300. During optimization analytical first and second derivatives were calculated in each step. The normal mode analysis was performed to secure only local minima of the multidimensional potential energy surface. After optimization, the G3MP2B3 composite method [11] was used to derive standard thermochemical properties. Subsequently, the enthalpy of formation, ΔfΗ°, was found using the atomization scheme. The relative Gibbs free energy values were then used to rank the thermodynamic stability of the thirty-five isomers.
Results and Discussion
29 out of the 35 calculated molecular structures have conventional chemical bonding while the remaining 6 are bimolecular complexes consisting of two molecular fragments. The fragments were small molecules such as CO, CO2, H2O2, O2, C2H2, C2H2O, CH2O, C3.
Structural comparison
Although, the structural generator software (Molgen 5.0) is expected to produce only tetravalent carbons, occasionally divalent carbene type carbon atoms have also appeared in some of the computed structures. This happened both in the open chained compounds as well as among the cyclic compounds. The structures of all of the 35 isomers are shown in Table 1 along with their thermodynamic data. It appears that that there are many ring systems among the isomers. There are five three membered rings, eight four membered rungs and six five membered rings. The clustering of the isomers in unimolecular structures and bimolecular complexes is shown in the entropy funnel representation in Figure 1, which illustrates the molecular network of stable isomers associated with the potential energy hyper-surface (PEHS) that describes the C3H2O2 empirical formula. The left-hand side of the double funnel collects all the unimolecular structures while the right hand side collects those consisting of two fragments. Only those isomers of the network can be joined by a line that may be directly inter-converted by a chemical reaction. For example structure #4 in Table 1 (H-CCCOOH) may undergo decarboxylation to form structure #1 ([CO2, H2C2])
| # | Isomer | ΔrelGº | S° | ΔfH° |
|---|---|---|---|---|
| 1 | [CO2,H2C2] | 0 | 352 | -175.3 |
| 2 | [CH2CO,CO] | 3.8 | 375 | -164.8 |
| 3 | 3-oxoacrylaldehyde |
23.4 | 297 | -168.3 |
| 4 | Propiolic acid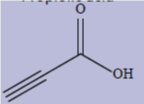 Data for the conformer where the OH group is oriented towards the CCH moiety |
81.9 | 298 | -109.6 |
| 5 | 2H-oxet-2-one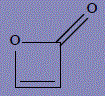 |
96.4 | 284 | -99.2 |
| 6 | 3-hydroxypropa-1,2-dien-1-one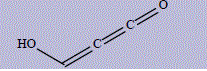 |
120.9 | 301 | -69.6 |
| 7 | 3-methylideneoxiran-2-one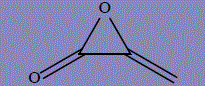 |
140.5 | 288 | -53.9 |
| 8 | 2-hydroxycycloprop-2-enone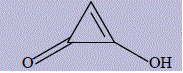 |
164.9 | 290 | -29.1 |
| 9 | ethynyl formate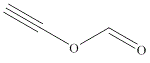 |
168.5 | 300 | -22.2 |
| 10 | 3-hydroxypropioaldehyde |
175.4 | 305 | -13.9 |
| 11 | cyclopropane-1,2-dione 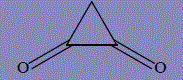 |
198.1 | 305 | 8.8 |
| 12 | 2-carbonyloxirane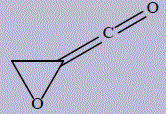 |
248.7 | 295 | 56.3 |
| 13 | prop-2-yn-3-id-1-ylidyneoxidanium hydrate  |
249.6 | 361 | 76.8 |
| 14 | 2-carboxyeth-1-en-1-ylidene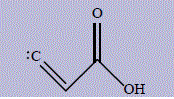 |
256 | 304 | 66.3 |
| 15 | 2H-1,3-dioxol-4-yl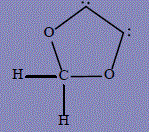 |
268.4 | 274 | 69.7 |
| 16 | (S)-2-formyl-2H-oxiren-1-ium-3-ide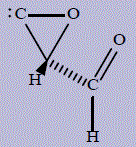 |
293.4 | 293 | 100.5 |
| 17 | 3,3-dihydroxyprop-1-yn-3-ylium-1-ide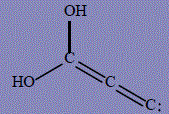 |
296.1 | 300 | 105.1 |
| 18 | cyclopropa-1(3),2-diene-1,2-diol |
302.3 | 291 | 108.8 |
| 19 | 2H-1λ4-oxet-1-ylium-3-olate, 3-oxo-3,4-dihydrooxet-1-ium-2-ide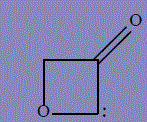 |
324.8 | 286 | 129.9 |
| 20 | 3-oxoprop-1-en-2-ylium-1-olate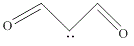 |
333.9 | 310 | 146 |
| 21 | 2-(methylidyne-λ4-oxy)ethen-1-one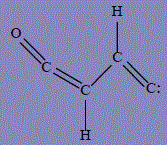 |
368.9 | 300 | 178 |
| 22 | (methylideneoxidaniumyl)ethynolate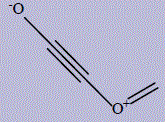 |
386.3 | 299 | 195.2 |
| 23 | (1R)-2-oxabicyclo[1.1.0]but-3-en-4-ol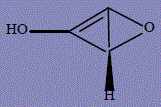 |
430.5 | 289 | 236.4 |
| 24 | (1r,3r)-2,4-dioxatricyclo[1.1.1.0¹,³]pentane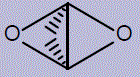 |
435.3 | 276 | 237.3 |
| 25 | 3-ethynyldioxirane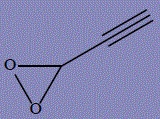 |
448.7 | 294 | 256.1 |
| 26 | 3H-1,2-dioxol-4-yl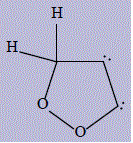 |
464.9 | 273 | 265.9 |
| 27 | [(3S)-3-hydroxyoxiran-2-ylidene]methylidene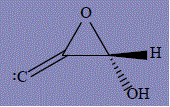 |
470.9 | 297 | 279.2 |
| 28 | 3-ethenylidenedioxirane or 3-vinylidenedioxirane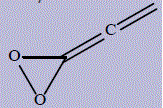 |
478.6 | 296 | 286.5 |
| 29 | [C2O,CH2O] | 572.9 | 337 | 393 |
| 30 | 1-hydroperoxycyclopropa-1,2-diene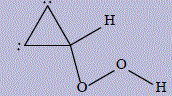 |
579.2 | 313 | 392.2 |
| 31 | Complex of dioxiren-1-ium-3-ide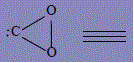 compound with acetylene |
582.2 | 381 | 415.6 |
| 32 | 1-[(1Z)-cycloprop-2-en-1-ylidene]dioxidan-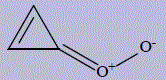 1-ium-2-ide 1-ium-2-ide |
613.2 | 294 | 420.5 |
| 33 | 3-(hydroxyoxidaniumylidene)prop-1-yn-1-ide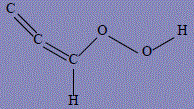 |
616.2 | 322 | 431.9 |
| 34 | [C3,H2O2] | 844.6 | 358 | 670.8 |
| 35 | [C3H2,1O2] | 846.1 | 356 | 672 |
Table 1: List of the investigated 35 isomers of C3H2O2. ΔrelG° (kJ/mol) is the standard free energy relative to that of the global minimum of [CO2, H2C2]. S° (J/(mol*K) is the standard entropy and ΔfH° (kJ/mol) is
the standard enthalpy of formation. ΔrelG° and S° were calculated at the G3MP2B3 level while ΔfH° was obtained using the atomization scheme.

Figure 1: Entropy-Funnel representation of thermodynamic properties of the 35 investigated C3H2O2 isomers. The global minimum (#1) and the propiolic acid (#4) are indicated with an arrows.
H-CC-COOH → [CO2, H2C2]
In that case structures #4 and #1, in Figure 1, may be interconnected by a linear, or curvy-linear, path that has a transition state somewhere along this line.
Thermodynamic stabilities
Since the propiolic acid is a well-studied compound one might expect that it was the most stable among the isomers, but it is not. The isomer with the lowest free energy (the global minimum) turns out to be a planar complex consisting of H2C2 and CO2. The complex has C2v symmetry. For convenience the stability of all isomers are listed relative to the global minimum as ΔrelGº. According to the G3MP2B3 calculations, propiolic acid exists in two conformations, differing in the orientation of the OH group relative to the CCH fragment. The conformer where the OH group is oriented away from the CCH fragment (conformer A) has a free energy which is 15 kJ/mol lower than the other isomer (conformer B). In Table 1, we list all the 35 isomers ranked according to ΔrelGº. Table 1 includes also ΔfHº and Sº along with an indication of the structure.
Transition states for interconversion
Topological properties of the potential energy hyper-surface: The C3H2O2 structure has seven atoms (n=7) implying the number of internal coordinates, 3n-6, is 15. This means that the PEHS has 15 independent variables:
E=f (X1,X2,X3…X14,X15)
The 35 minima are located on the PEHS, however, without explicit knowledge of the topology of the hyper-surface it is impossible to know which pair of minima is adjacent and which ones are further separated from each other. Those that are adjacent have a transition state in between to enhance the conversion of one minimum to another. Other non-adjacent minima can only be converted through multiple conversions involving more than one transition state.
case study for interconversion: The transition from the global minimum, [CO2, C2H2], to the propionic acid isomer has been determined using the QST2 method implemented in Gausian09 [12]. The transition state was determined for both conformers of propiolic acid. It turns out that the two transition states are identical. Investigation of the transition state using the internal reaction coordinate method [12] shows that it connects the global minimum to the conformer, where the OH group is oriented towards the CCH moiety of propionic acid (conformer B). The structure of the transition state is depicted in Figure 2. The free energy barrier for this transition at standard condition is 321 kJ/mol at the G3MP2B3 level. The barrier is so high that the reaction of CO2, C2H2], to the propionic acid isomer has been determined usin and C2H2 to form propionic acid will not happen - in line with observations. Calculations of the transition state using other levels of theory lead to similar estimates for the barrier height as reported in Table 2. Conformer B of propionic acid is connected to conformer A (which has lower free energy). A QST2 calculation leads to an estimate for the barrier height of 32 kJ/ mol (G3MP2B3). Thus the transition from the global ground state to the most stable isomer of propionic acid is a two state process.

Figure 2: Transition state connecting the global minimum, [CO2, C2H2], among the C3H2O2 isomers with the propiolic acid isomer, HCC-COOH, in a conformation where the OH group is oriented towards the CCH moiety. The transition state is calculated at the G3MP2B3 level of theory. The angle O2-C1-O3 is 144°. The distances 7H-2O and 7H-5C are 1.32 Å and 1.33 Å, respectively. The structure of the transition state is planar.
| Level of theory | E1> | E2 |
|---|---|---|
| B3LYP/6-31G(d) | 298 | 33.1 |
| MP2/6-31G(d) | 337 | 33.1 |
| WB97XD/AUG-cc-pVDZ | 297 | 32.7 |
| G3MP2B3 | 321 | 32.1 |
Table 2: Barrier heights (kJ/mol) for the transition from the global minimum, [CO2, C2H2], to propiolic acid calculated at various levels of theory. E1 is the barrier from the global minimum to conformer B of propionic acid and E2 is the barrier from conformer B to conformer A.
Conclusion
It was shown that new molecular structures can be predicted by molecular computations and the relative stabilities can be assessed by computing thermodynamic functions. Even though this is a small step on a long road, it is hoped that such research would be useful in designing new drugs and other desirable molecules.
References
- Elamari H , Slime R, Chabot GG, Quentin L, Scherman D, et al.(2013) Synthesis and in Vitro Evaluation of Potential Anticancer Activity of Mono- and Bis-1,2,3-Triazole Derivatives of Bis-Alkynes. Eur J Med Chem 60: 360-364.
- Haritos VS, Horne I, Damcevski K, Glover K, Gibb N, et al. (2012) The Convergent Evolution of Defensive Polyacetylenic Fatty Acid Biosynthesis Genes in Soldier Beetles. NatCommun 3:1150.
- Karpi?ska G,Mazurek AP,Dobrowolski JC (2011) Hydroxyquinolines: Constitutional Isomers and Tautomers. Comput. Theor Chem 972:48-56.
- Khriachtchev L (2008) Rotational Isomers of Small Molecules in Noble-Gas Solids: From Monomers to Hydrogen-Bonded Complexes. J Mol Struct 880:14-22.
- Li X-C, Jacob MR, Khan SI, Ashfaq MK, Babu KS, et al. (2008) Potent in Vitro Antifungal Activities of Naturally Occurring Acetylenic Acids. Antimicrob agents chemother 52: 2442-2448.
- Valadbeigi Y, Farrokhpour H(2013)DFT, CBS-Q, W1BD and G4MP2 calculation of the proton and electron affinities, gas phase basicities and ionization energies of saturated and unsaturated carboxylic acids (C1-C4),Int J Quantum Chem 113: 1717-1721.
- Li J, Brill TB (2002)Spectroscopy of Hydrothermal Reactions 20: Experimental and DFT Computational Comparison of Decarboxylation of Dicarboxylic Acids Connected by Single, Double, and Triple Bonds. J Phys Chem A 106: 9491-9498.
- Lewis RJ (2004) Sax's Dangerous Properties of Industrial Materials. 11th edn. Wiley-Interscience, Wiley & Sons, Inc. Hoboken, NJ. USA. p: 3069.
- Riemenschneider W (2008) Ullmann's Encyclopedia of Industrial Chemistry. 7th edn.New York, NY. John Wiley & Sons; Carboxylic Acids, Aliphatic.
- Benecke C, Grüner T,Kerber A,Laue R,Wieland T (1997) MOLecular structure GENeration with MOLGEN, new features and future developments. Fresenius' J Anal Chem 359:23-32.
- Baboul AG, Curtiss LA, Redfern PC, Raghavachari K (1999) Gaussian-3 theory using density functional geometries and zero-point energies. J Chem Phys 110:7650-7657.
- Frisch MJ(2009) Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford, CT, USA.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences